Tema: Bioinformatic methods for integrating whole-genome expression results into cellular networks
Autores: Duccio Cavalieri and Carlotta De Filippo
Introducción
Este artículo muestra como se realiza una crítica a los métodos de técnicas que se utilizan para realizar una conexión de la información de estudios sobre genómica funcional a la función biológica.
Se discuten los métodos de análisis del clúster para inferir en correlación entre los genes, como son los métodos para la integración de la información de genes con la información existente en las vías biológicas y los métodos que combinan análisis de conglomerados con la información biológica para reconstruir redes biológicas novedosas.
La regulación de la expresión génica y la actividad de la proteína son esenciales para la función de los sistemas moleculares y celulares.
Algunos métodos de alto rendimiento ofrecen diversos puntos de vista de los genes que participan y cada vez es más claro los análisis globales en conocimiento representa un desafío sin precedentes.
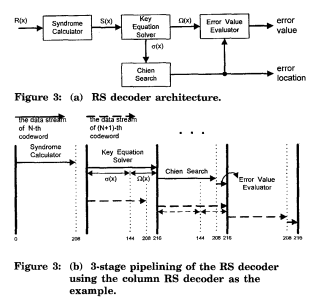
Se describen tres principales enfoques de bioinformáticas, como se muestra en la Figura 1:

Métodos que utilizan
la información genómica para inferir correlaciones entre los genes y grupos de
genes
Es difícil interpretar datos de microarrays de un grupo de
genes, que se encuentran sobre la base de la regulación y funciones similares o estado similar
celular y el fenotipo biológico.
Este es un problema ha demostrado ser atractivo para bioestadísticos, de modo que en los últimos años el análisis estadístico ha sido uno de los campos de investigación más activas de microarrays.
La identificación de clases desconocidas de genes coregulados utilizando perfiles de expresión de todo el genoma o la clasificación en clases conocidas de genes funcionalmente relacionadas son dos técnicas comunes que se utilizan en experimentos de expresión génica.
Este es un problema ha demostrado ser atractivo para bioestadísticos, de modo que en los últimos años el análisis estadístico ha sido uno de los campos de investigación más activas de microarrays.
La identificación de clases desconocidas de genes coregulados utilizando perfiles de expresión de todo el genoma o la clasificación en clases conocidas de genes funcionalmente relacionadas son dos técnicas comunes que se utilizan en experimentos de expresión génica.
La agrupación de los algoritmos basados en modelos de
probabilidad son métodos en donde los datos sean generados por una mezcla de
distribuciones de probabilidades.
Una característica de la mezcla de modelado es que las probabilidades
posteriores pertenecen a una clase.
En la investigación en el análisis se utilizaron gráficas de
modelado de Gauss, con datos informativos confiables de subconjuntos, basados
en la agrupación de la descomposición de valor singular, teoría de grafos,
mapas auto organizados simuladas y las redes libres de escala.
Algunos métodos de la agrupación, desarrollados para identificar
subconjuntos de genes pueden ser supervisados mediante el uso de propiedades conocidas
de los genes para ayudar a encontrar asociaciones significativas.
Todos estos métodos se basan en la suposición de los genes relacionados
tienen niveles similares de expresión génica, la suposición de que podría
significar que los genes importantes se pasan por alto.
Métodos que integran
resultado de la genómica funcional en las vías metabólicas o celulares
Las redes bioquímicas enlazan las enzimas del flujo de
substratos y los productos de las diferentes reacciones, las redes reguladoras
describen cómo se regula la expresión de los genes que codifican las enzimas de
las vías; redes de interacción proteína-proteína anotar las proteínas que
interactúan.
Una de las principales diferencias entre una red y un camino,
es que una vía es un conjunto ligado de reacciones bioquímicas, e incluye la
idea de direccionalidad, donde las líneas se convierten en flechas, y contienen
información sobre el flujo de energía y el metabolito.
Por lo tanto, varias iniciativas han abordado el análisis de
datos de microarrays en el contexto del conocimiento de la técnica de las redes
biológicas o caminos.
Existen métodos de análisis de microarrays basados en vías que
buscan patrones de expresión en las clases predefinidas de genes, tales como
los implicados en el metabolismo, el control de la división celular, la
apoptosis, el transporte de membrana, la reproducción sexual, de señalización,
y así sucesivamente, con el objetivo de integrar la información obtenida en una
escala genómica con la información biológica.
La integración de la información genómica es cada vez más
importante con la aparición de la "biología de sistemas". El análisis
de los diferentes tipos de datos genómicos se compromete a fomentar un nuevo
nivel de comprensión del sistema en su conjunto.
Los esfuerzos por establecer la ontología de genes se están
convirtiendo cada vez más importante con el avance de los diversos proyectos de
secuenciación del genoma y son pertinentes para la interpretación de los datos
genómicos en su contexto biológico.
La base de datos de GO proporciona una herramienta útil para
anotar y analizar las funciones de un gran número de genes, sa disponibilidad envía
una indicación adecuada y que es uno de los requisitos para el análisis de
datos de microarrays en el contexto de las vías biológicas.
La información contenida en esta base de datos es
proporcionada por biólogos que son expertos en un dominio específico de la
biología, editado y que se cruza con PubMed, IR, y las bases de datos de
secuencias como pueden ser LocusLink, Ensembl y SwissProt, es revisada por otros investigadores
biológicos para la consistencia y precisión y se hará pública, la información es paralela al desarrollo de un formato de
intercambio de datos, la vía de la información es esencial para compartir,
evaluar y desarrollar los recursos de información vía y modelos basados en
vía.
SBML es un formato para la representación de modelos de redes de reacciones bioquímicas, como las redes metabólicas, da un apoyo para la integración de diferentes herramientas de modelado y simulación, este lenguaje es muy detallado y describe específicamente las variables biológicas.
La principal diferencia entre BioPAX y SBML, es que BioPAX es esencialmente un formato para el intercambio de información entre diferentes bases de datos, mientras que SBML es un soporte para el modelado y la simulación.
Los esfuerzos actuales para desarrollar enfoques basados para el análisis de datos de microarrays se pueden dividir en dos clases principales: en primer lugar, los métodos que muestran automáticamente los resultados de la genómica funcional, y segundo, los métodos de prueba para la significación estadística de enriquecimiento de los genes.
Métodos que muestran automáticamente los resultados de la genómica funcional en las cartas metabólicos o celular
Un itinerario de ruta es un diagrama que muestra las relaciones biológicas entre los genes o productos génicos basados en principios de organización, tales como las vías metabólicas, cascadas de transducción de señales o localizaciones sub-celulares.
Algunos análisis de microarrays comerciales como ResolverTM Rosetta, GeneSpringTM, AcuityTM, GeneGOTM y BIOKNOWLEDGE LibraryTM han desarrollado características que habilitan la visualización de datos de expresión génica en el contexto de mapas metabólicos, genéticos o interacción.
GeneMapp es uno de las aplicaciones informáticas más interesantes que se disponibles libremente, está diseñado para visualizar la expresión génica global u otros tipos de datos genómicos en el contexto de cientos de MAPPS las vías existentes y miles de términos para Gene Ontología, y facilita el intercambio de datos vía relacionadas entre investigadores.
MAPP es un formato de archivo especial, asignando a cada gen una identificación (ID) tomada de GenBank, o un sistema de ID definido por el usuario, Es independiente de los datos de expresión génica y del principio de organización, y permite la visualización de las vías conocidas de bases de datos curada o la construcción de MAPPS de acuerdo con los criterios definidos por el usuario, sin necesidad de especificar el número y el tipo de interacciones entre los elementos.
Otra herramienta es GenMAPP, proporciona una característica adicional única en cuanto a los recursos de la vía existente, ya que permite al usuario modificar las vías para su propio uso o para el diseño de nuevas vías, es una herramienta poderosa para el intercambio de datos vía libremente relacionados entre los investigadores.
Cytoscape es una herramienta útil para la integración de datos genómicos en redes de genes, esta herramienta de código abierto permite la visualización, dibujo y edición de las redes de interacción molecular.
Métodos de prueba
para estadísticas de enriquecimiento de los genes pertenecientes a la misma
clase, vía, o red.
El desarrollo de métodos estadísticos para evaluar la
importancia de la alteración en la expresión de diversas vías celulares es de
mucho interés.
Varios trabajos de microarrays han informado la activación o
represión de un camino dado, pero con demasiada frecuencia el investigador se encuentra
lo que ya sabe.
Para evaluar la importancia de los genes de una vía que será
cambiado de forma coordinada en la expresión en un experimento dado, se tienen
que tomar varios factores: el número de marcos de lectura abierta para los que
se ha alterado la expresión en cada vía, el número total de ORFs contenida en
la vía, la proporción de los ORFs en el genoma en una determinada vía, y la
correlación de las vías, para seleccionar la prueba estadística más apropiada.
El programa analizador “Camino de Procesador” utiliza la
prueba exacta de Fisher para generar un valor de “p” que indica la probabilidad
de que la vía podría contener tantos o más afectada genes que realmente
observados.
La clasificación para la prueba exacta de Fisher puede
utilizarse para comparar diferentes experimentos, la comparación se puede
representar gráficamente utilizando programas tan comunes como ExcelTM o
TreeView, o los más sofisticados, como OpenDX.
La práctica común sugiere que la validez de los métodos
estadísticos se ha probado en un problema biológico conocido.
Existen varios métodos para evaluar la importancia de los
cambios de expresión de diversas vías celulares se han desarrollado
recientemente.
Uno de esos métodos es GeneMerge, utiliza listas de genes de
KEGG, GO, MIPS o de otras fuentes, y el rango calificaciones de
sobre-representación funcional o categórica en el estudio conjunto de genes y
se obtiene utilizando la distribución hipergeométrica, el programa es muy útil,
ya que se extiende el análisis a cualquier lista de favoritos de los genes, las
principales limitaciones son la ausencia de cualquier forma de visualización
vía.
Otro método es GOstat, también tiene aportaciones de todos
los genes en un microarray y obtiene automáticamente los GO anotaciones de una
base de datos, y genera estadísticas de
los cuales las anotaciones están excesivamente representadas en la lista de los
genes analizados.
“Camino minero” es otra herramienta de libre acceso que
utiliza la prueba exacta de Fisher para clasificar los genes que están
definidas como parte de la misma vía, sobre la base de su papel en las vías
metabólicas, celular y regulador, o como grupos pre-definidos por los el
usuario.
Una de las principales limitaciones de estos métodos es la
aplicación de la prueba exacta de Fisher de la distribución hipergeométrica
para el análisis de las vías altamente interconectadas con un alto nivel de
redundancia.
La integración de la información de análisis de la expresión
basada en las vías con el análisis de flujo de equilibrio se puede convertir en
modelos de regulación que se pueden combinar además con modelos genoma escala
metabólica para construir modelos integrados de la función celular, incluyendo
tanto el metabolismo y su regulación.
Por otro lado, será interesante ver los resultados de la
aplicación de redes bayesianas para la representación de las dependencias
estadísticas para el descubrimiento de las interacciones entre los genes en las
vías y descubrir nuevas vías, de acuerdo con el mismo marco que se utiliza para
describir las interacciones entre los genes.
Métodos que utilizan
los resultados funcionales de la genómica, información biológica existente, y
la agrupación de reconstruir una red biológica novela
Uno de los límites de los métodos basados en la vía es que
no proporcionan información sobre los genes de función desconocida.
La integración de los resultados de los análisis basados
en la vía con los de los algoritmos de agrupamiento podría indicar los genes
de función junto con genes asignados a una determinada vía, lo que sugiere que
sus funciones son metabólicamente relacionados, y que ofrezcan un nuevo enfoque
para la atribución de funciones a genes desconocidos.
GenExpress es una herramienta desarrollada recientemente que
permite al usuario combinar análisis conglomerados con el análisis de atributos
biológicos.
Esta herramienta se ha aplicado recientemente a la colección
de datos de expresión disponibles relacionados con la progresión del cáncer, lo
que demuestra una mina muy prometedora de la información biológica y clínica.
Conclusiones
La ventaja de analizar una red de genes es que los genes
alterados que varían pueden ser considerados significativos, en lugar de
centrarse en la probabilidad de que el cambio de cada elemento.
La hipótesis de que los genes en la misma vía son más
propensos a ser regulados de manera coordinada.
Los métodos que analizan los datos de expresión de acuerdo a
una lógica basada en las vías deben dar una indicación de la importancia estadística
de las conclusiones, proporcionar una interfaz fácil de usar para la
visualización de los resultados, ser capaz de abarcar el mayor número de vías
bien establecidas, definir nuevas vías, asignar los genes desconocidos para una
vía, y reconstruir la estructura jerárquica de un grupo de vías.
El desarrollar formatos de intercambio comunes para las vías
biológicas, anotaciones de todas las reacciones bioquímicas y bases de datos de
vías, son fundamentales para la integración de la función bioquímica y la
expresión génica.
El mejorar los métodos basados en la vía será necesario
mejorar las estadísticas, la conexión de las vías biológicas con información
sobre los factores de transcripción, el flujo de metabolitos, redes de
interacción proteína-proteína, y una mejor visualización y herramientas gráficas.
Los avances en este campo será una contribución fundamental
para la aplicación de microarrays en los
estudios clínicos y la biología de sistemas.
El en la investigación mencionaban mucho el utilizar bases de datos y software como métodos para realizar investigaciones sobre genes y moléculas, cabe mencionar que hoy en día, se pueden utilizar herramientas mas potentes para realizar investigaciones mas exactas.
El combinar nuevas tecnologías para realizar chequeo de genes e investigaciones, es algo demasiado complicado, aunque en estos tiempos modernos, el realizar este tipo de proyectos es muy cotizado.
Referencia:
- Bioinformatic methods for integrating whole-genome expression results into cellular networks, Duccio Cavalieri and Carlotta De Filippo [PDF], pages: 727 - 734.
Publicado en: Drug Discovery Today, Volume 10 (10) Elsevier - May 15, 2005
Tipo de producto: Publicacion de Revista
Disponible en: http://www.deepdyve.com/lp/elsevier/bioinformatic-methods-for-integrating-whole-genome-expression-results-oARbqZXgyx
Ultimo acceso: 28/05/2013